



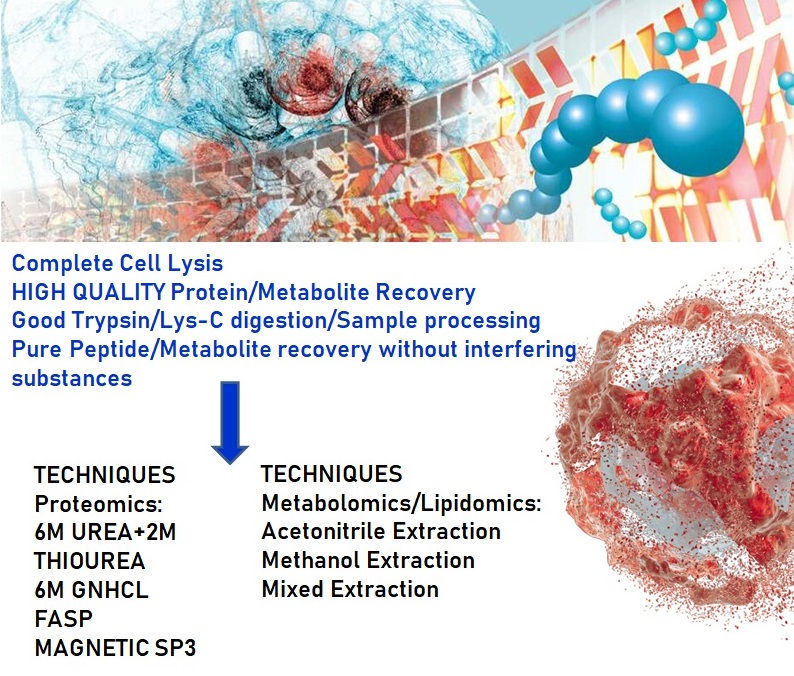
HIGH QUALITY SAMPLE PREP
FOR BETTER MS ANALYSIS
Proper sample preparation for MS-based analysis is a critical step in the Mass Spectrometry workflow because it can be both variable and time consuming. The quality and reproducibility of sample extraction and preparation significantly impacts MS results. Our Laboratory recognize that sample preparation, instrumentation and software are all critical to Mass Spectrometry based research success; therefore, all three components must be properly integrated into robust workflows for consistent, high quality results.
At Vproteomics we take care of the client project by preparing high quality peptides/proteins/metabolites in our Mass Spectrometry facility.
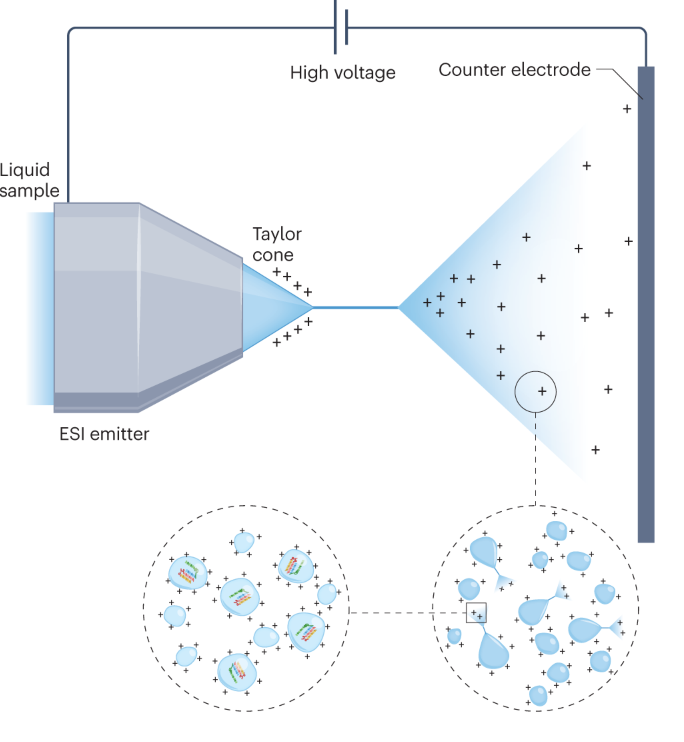
LATEST CHROMATOGRAPHY SETUP
FOR BETTER SPRAY
Using the best available UHPLC chromatography columns with temperature controlled setup we ensure that the peptide samples are well separated before entering the Mass Spectrometer.
Extra fine tip nanocolumns ensure the highest possible performance, combining powerful chemistries with a column architecture that eliminates post-column loss.

REPRODUCIBLE CHROMATOGRAPHY SETUP
ACCURATE LFQ QUANT RESULTS
Unbiased proteomic studies can be performed using our high resolution Orbitrap mass spectrometers to provide accurate quantification data. We provide Label-free methods for both relative and absolute quantification, which are rapid and a low-cost alternative to other quantitative proteomic approaches.
For differential proteomics analysis experiments, we generally recommend 3-4 biological replicates each condition to result in proper statistical analysis.
QC Peptides Multiple replicates
LATEST MASS SPECTROMETER
BEST RESULTS
Orbitrap Exploris:
“Most advanced mass spectrometer for the most advanced research applications”
Excel at the most challenging of applications, including low level PTM analysis, multiplexed relative quantitation using isobaric tags, intact protein characterization, with the latest Exploris Mass Spectrometer.
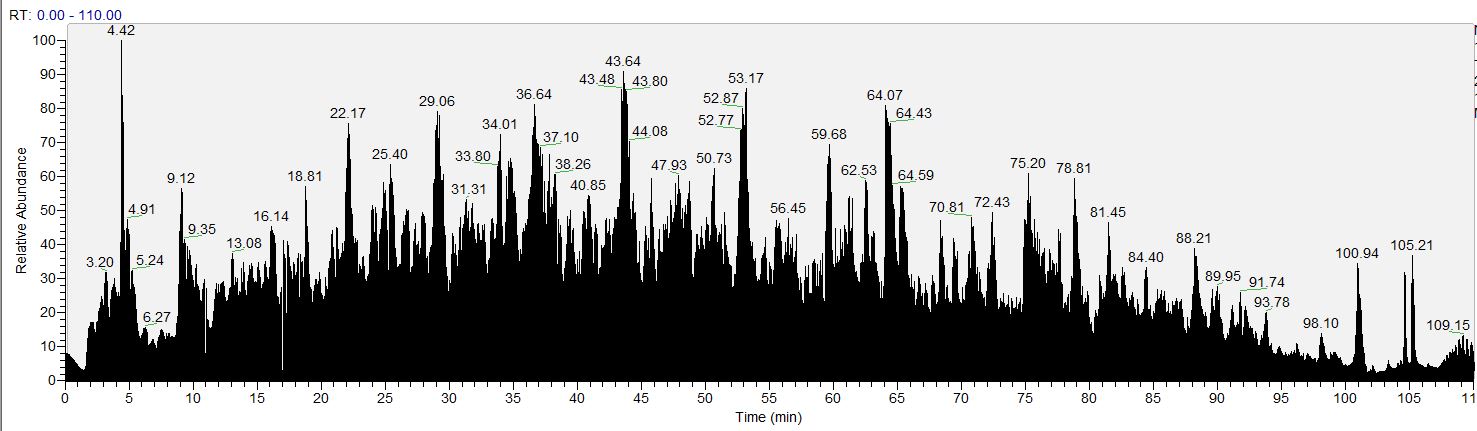

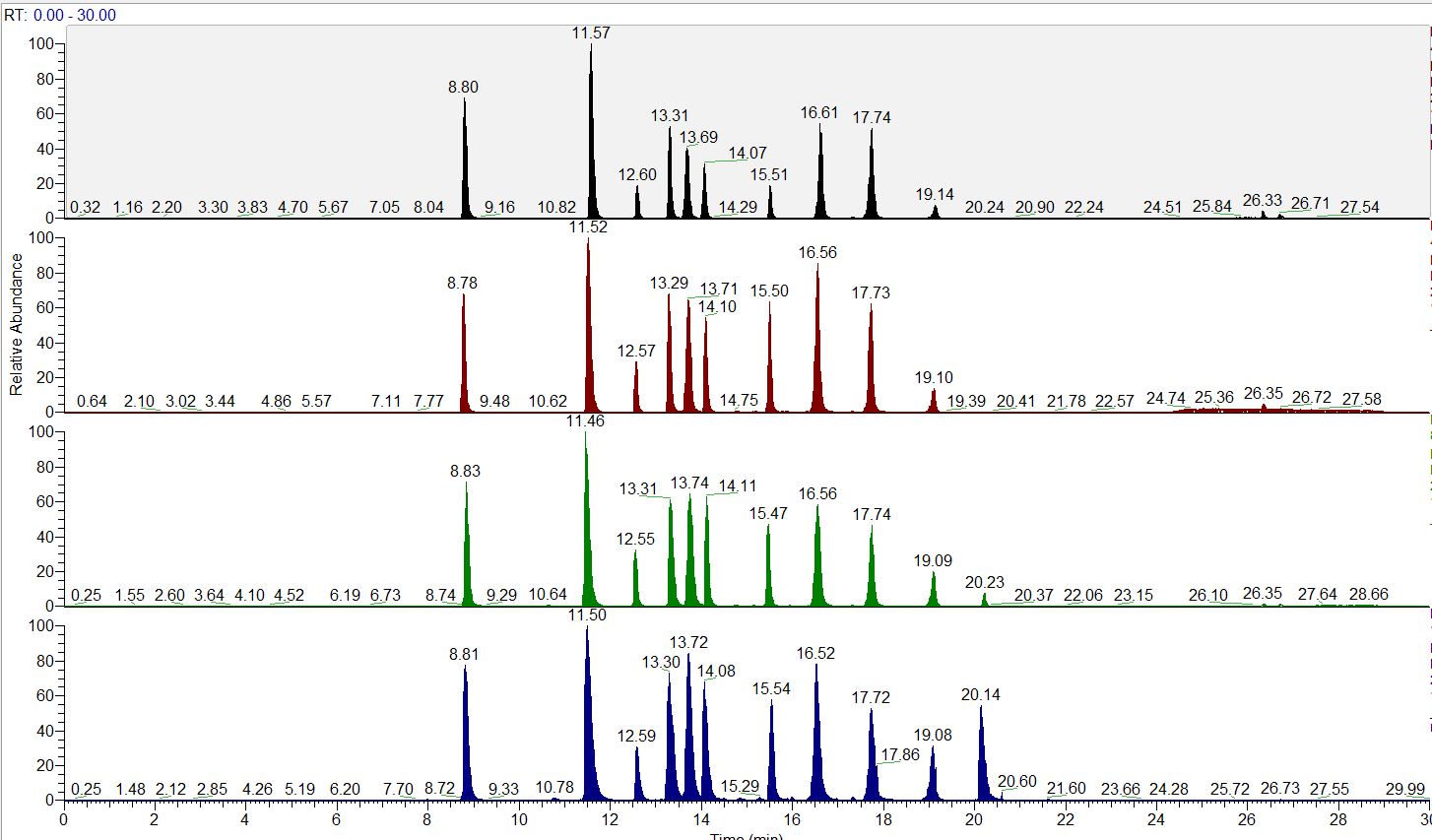
HIGH THROUGHPUT DISCOVERY METABOLOMICS
DIFFERENT CHEMISTRY FOR DIFFERENT PROJECTS
Quantitation
- Advanced quadrupole technology (AQT) improves precursor selection and transmission for more accurate quantitation of low-abundance analytes in complex matrices.
- Sophisticated data-independent acquisition (DIA) and parallel reaction monitoring (PRM) deliver reproducible quantitation with complete qualitative confidence.
- An advanced active beam guide (AABG) reduces noise and extends maintenance intervals.
High-Confidence Results
- HR/AM and full-scan capabilities capture all sample data, all of the time, enabling retrospective data analysis without need to re-run samples.
- Resolving power up to 140,000 FWHM eliminates isobaric interferences, increasing confidence in results when analyzing samples.
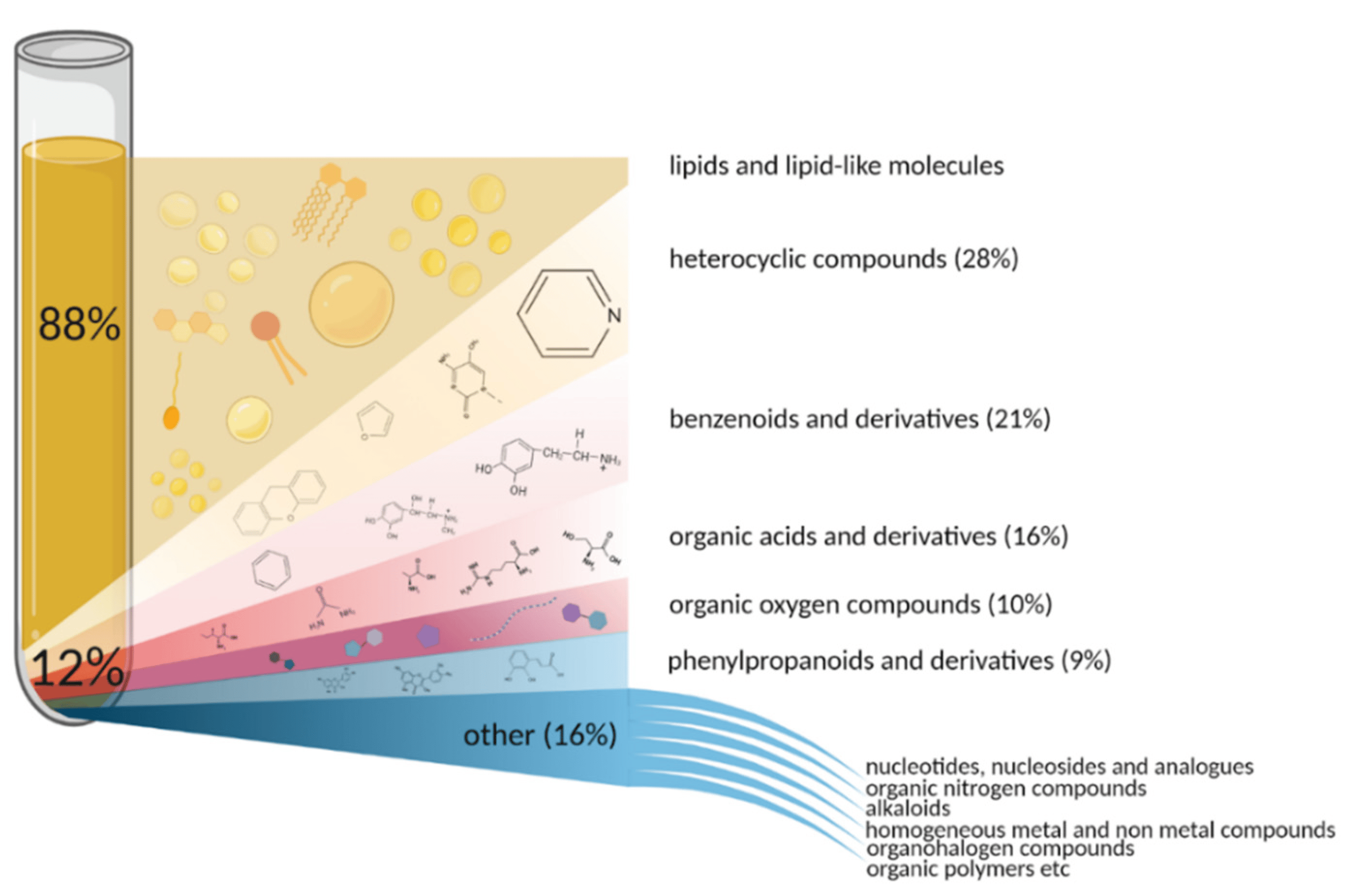
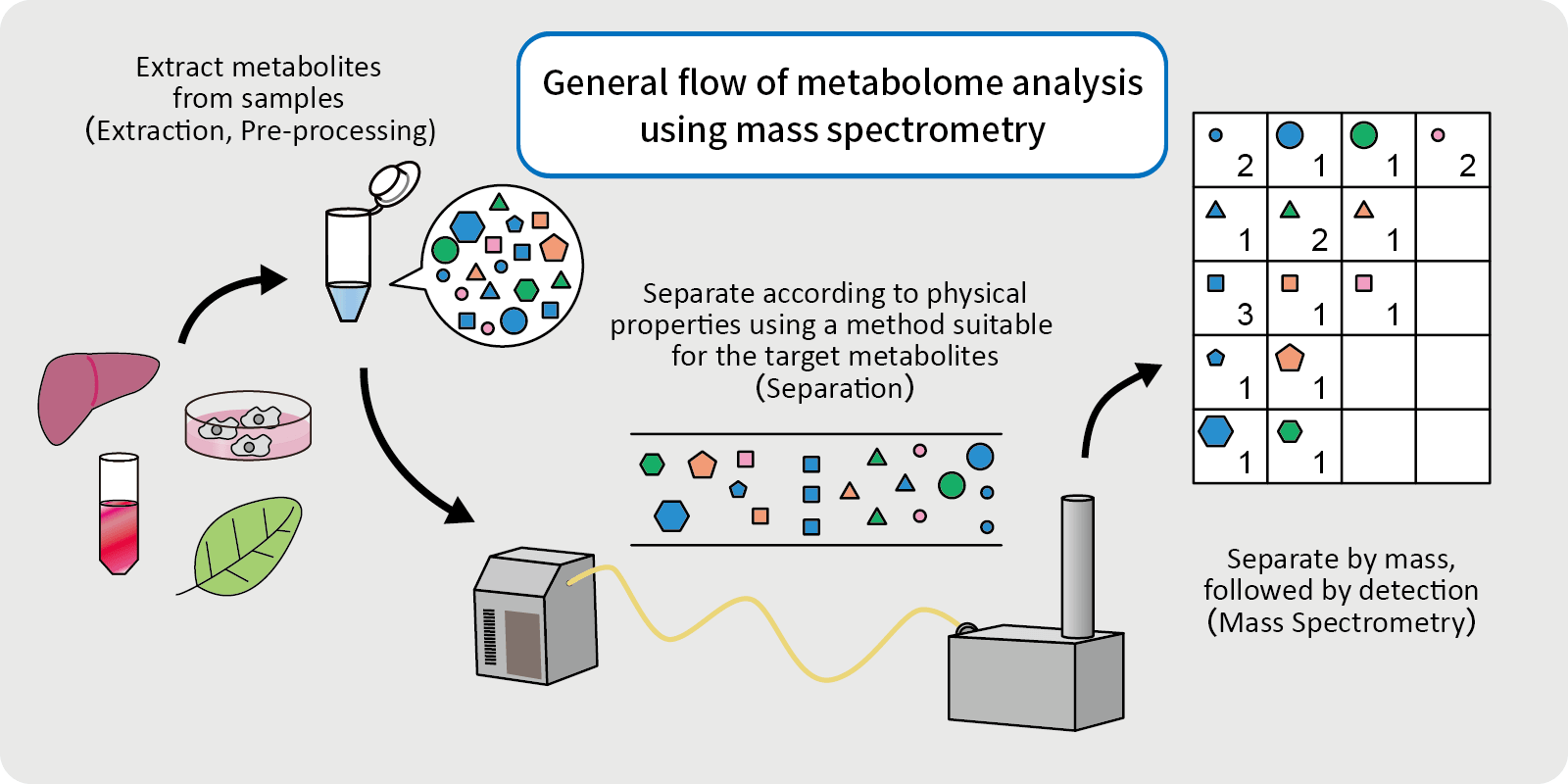
HIGH POWER COMPUTING INFRASTRUCTURE
CUSTOMIZED PIPELINES FOR DIFFERENT PROJECTS
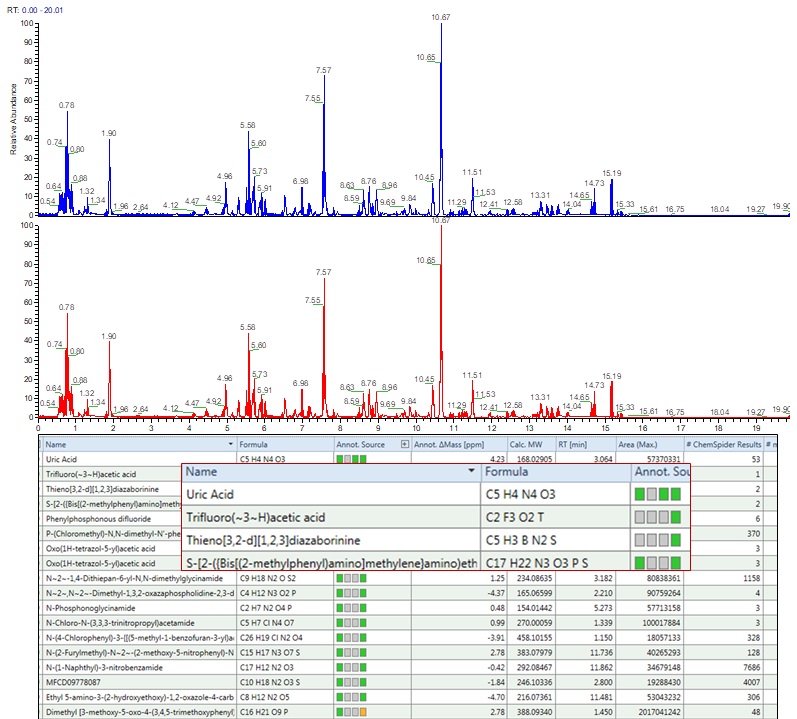
Advanced Data Analysis Pipelines
- Standardized pipelines for high resolution raw data analysis for annotation of identified proteins and other molecules
- Usage of both commercial and open-source tools for results compilation
- Routine up-gradation of bioinformatics tools for Mass Spec data mining.
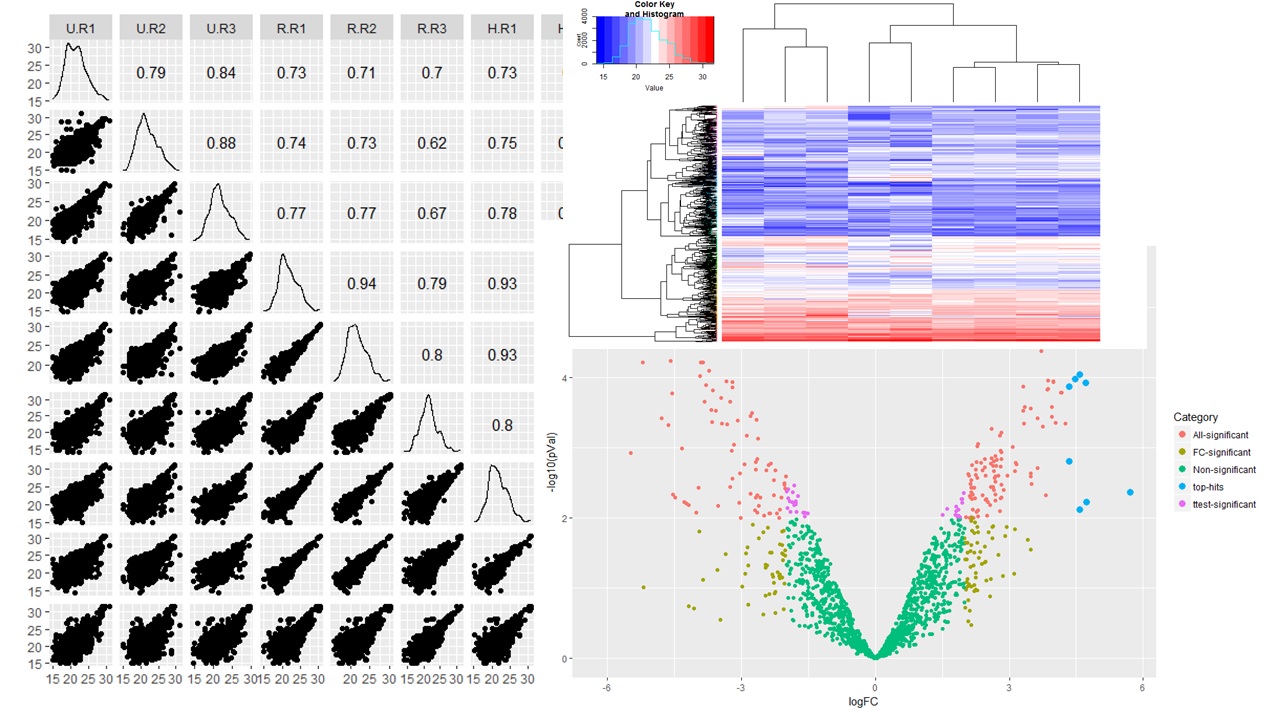
High-Confidence Statistical Analysis
- Support to the clients in designing the Mass Spec experiment right from the beginning to get good power anaysis
- Results provided with relevant statistical tests with significant and non-significant hits.
- High quality data analytics support depending upon experimental requirements.
- Support for raw data submission in OMICS data repositories